YamboConvergence: automated convergence¶
The YamboConvergence workchain provides the functionalities to run multiple G0W0 calculations on the same system, over a wide range of chaging parameter.
This represents the typical method to obtain an accurate evaluation of the quasiparticle correction: indeed, a lot of effort has to be done in order to find
the convergence with respect to parameters like empty states used to evaluate the Self Energy used to solve the quasiparticle equation.
If you implement a logic for convergence evaluation to guide the iter of these multiple parameter-dependent calculations, you can obtain an automatic flow
that allows you to obtain good G0W0 results. This is what YamboConvergence does, in its ‘1D_convergence’ mode. The philosophy behind is the iteration over a single
parameter at the time, choosing the cheapest converged result as starting point eahc time you change the interested parameter. A lot of effort was put in order to have a more flexible procedure to run multiple calculations, focusing on a very generic convergence
scheme: setup, run a group of calculations, check the results, stop or do other calculations. Repeat. This allowed us to define different logic to perform
convergence analysis by simply enriching the python functions and classes imported by the workflow. For example, in the future it will be possible to evaluate
convergence also by performing fittings over the parameters, defining by hand (case-sensitive) the fitting function. For now, we just evaluate the convergence
by evaluating the difference between the last calculation done and the previous ones, with respect to a given threshold (like tens of meV). This threshold has
to be provided in the workflow_settings, as you can see from the example.
Let’s see the case of automatic search of convergence over an arbitrary number of parameters, activated by setting "type": 1D_convergence:
Example usage:
#!/usr/bin/env runaiida
# -*- coding: utf-8 -*-
from __future__ import absolute_import
from __future__ import print_function
import sys
import os
from aiida.plugins import DataFactory, CalculationFactory
from aiida.orm import List, Dict
from aiida.engine import submit
from aiida_yambo.workflows.yamboconvergence import YamboConvergence
from aiida_quantumespresso.utils.pseudopotential import validate_and_prepare_pseudos_inputs
from ase import Atoms
import argparse
def get_options():
parser = argparse.ArgumentParser(description='YAMBO calculation.')
parser.add_argument(
'--yambocode',
type=int,
dest='yambocode_pk',
required=True,
help='The yambo(main code) codename to use')
parser.add_argument(
'--parent',
type=int,
dest='parent_pk',
required=False,
help='The parent to use')
parser.add_argument(
'--yamboprecode',
type=int,
dest='yamboprecode_pk',
required=True,
help='The precode to use')
parser.add_argument(
'--pwcode',
type=int,
dest='pwcode_pk',
required=True,
help='The pw to use')
parser.add_argument(
'--pseudo',
type=str,
dest='pseudo_family',
required=True,
help='The pseudo_family')
parser.add_argument(
'--time',
type=int,
dest='max_wallclock_seconds',
required=False,
default=30*60,
help='max wallclock in seconds')
parser.add_argument(
'--nodes',
type=int,
dest='num_machines',
required=False,
default=1,
help='number of machines')
parser.add_argument(
'--mpi',
type=int,
dest='num_mpiprocs_per_machine',
required=False,
default=1,
help='number of mpi processes per machine')
parser.add_argument(
'--threads',
type=int,
dest='num_cores_per_mpiproc',
required=False,
default=1,
help='number of threads per mpi process')
parser.add_argument(
'--queue_name',
type=str,
dest='queue_name',
required=False,
default=None,
help='queue(PBS) or partition(SLURM) name')
parser.add_argument(
'--qos',
type=str,
dest='qos',
required=False,
default=None,
help='qos name')
parser.add_argument(
'--account',
type=str,
dest='account',
required=False,
default=None,
help='account name')
args = parser.parse_args()
###### setting the machine options ######
options = {
'yambocode_pk': args.yambocode_pk,
'yamboprecode_pk': args.yamboprecode_pk,
'pwcode_pk': args.pwcode_pk,
'pseudo_family': args.pseudo_family,
'max_wallclock_seconds': args.max_wallclock_seconds,
'resources': {
"num_machines": args.num_machines,
"num_mpiprocs_per_machine": args.num_mpiprocs_per_machine,
"num_cores_per_mpiproc": args.num_cores_per_mpiproc,
},
'custom_scheduler_commands': u"export OMP_NUM_THREADS="+str(args.num_cores_per_mpiproc),
}
if args.parent_pk:
options['parent_pk']=args.parent_pk
if args.queue_name:
options['queue_name']=args.queue_name
if args.qos:
options['qos']=args.qos
if args.account:
options['account']=args.account
return options
def main(options):
###### setting the lattice structure ######
alat = 2.4955987320 # Angstrom
the_cell = [[1.000000*alat, 0.000000, 0.000000],
[-0.500000*alat, 0.866025*alat, 0.000000],
[0.000000, 0.000000, 6.4436359260]]
atoms = Atoms('BNNB', [(1.2477994910, 0.7204172280, 0.0000000000),
(-0.0000001250, 1.4408346720, 0.0000000000),
(1.2477994910, 0.7204172280, 3.2218179630),
(-0.0000001250,1.4408346720, 3.2218179630)],
cell = [1,1,1])
atoms.set_cell(the_cell, scale_atoms=False)
atoms.set_pbc([True,True,True])
StructureData = DataFactory('structure')
structure = StructureData(ase=atoms)
###### setting the kpoints mesh ######
KpointsData = DataFactory('array.kpoints')
kpoints = KpointsData()
kpoints.set_kpoints_mesh([6,6,2])
###### setting the scf parameters ######
Dict = DataFactory('dict')
params_scf = {
'CONTROL': {
'calculation': 'scf',
'verbosity': 'high',
'wf_collect': True
},
'SYSTEM': {
'ecutwfc': 130.,
'force_symmorphic': True,
'nbnd': 20
},
'ELECTRONS': {
'mixing_mode': 'plain',
'mixing_beta': 0.7,
'conv_thr': 1.e-8,
'diago_thr_init': 5.0e-6,
'diago_full_acc': True
},
}
parameter_scf = Dict(dict=params_scf)
params_nscf = {
'CONTROL': {
'calculation': 'nscf',
'verbosity': 'high',
'wf_collect': True
},
'SYSTEM': {
'ecutwfc': 130.,
'force_symmorphic': True,
'nbnd': 500
},
'ELECTRONS': {
'mixing_mode': 'plain',
'mixing_beta': 0.6,
'conv_thr': 1.e-8,
'diagonalization': 'david',
'diago_thr_init': 5.0e-6,
'diago_full_acc': True
},
}
parameter_nscf = Dict(dict=params_nscf)
KpointsData = DataFactory('array.kpoints')
kpoints = KpointsData()
kpoints.set_kpoints_mesh([6,6,2])
alat = 2.4955987320 # Angstrom
the_cell = [[1.000000*alat, 0.000000, 0.000000],
[-0.500000*alat, 0.866025*alat, 0.000000],
[0.000000, 0.000000, 6.4436359260]]
atoms = Atoms('BNNB', [(1.2477994910, 0.7204172280, 0.0000000000),
(-0.0000001250, 1.4408346720, 0.0000000000),
(1.2477994910, 0.7204172280, 3.2218179630),
(-0.0000001250,1.4408346720, 3.2218179630)],
cell = [1,1,1])
atoms.set_cell(the_cell, scale_atoms=False)
atoms.set_pbc([True,True,True])
StructureData = DataFactory('structure')
structure = StructureData(ase=atoms)
params_gw = {
'HF_and_locXC': True,
'dipoles': True,
'ppa': True,
'gw0': True,
'em1d': True,
'Chimod': 'hartree',
#'EXXRLvcs': 40,
#'EXXRLvcs_units': 'Ry',
'BndsRnXp': [1, 10],
'NGsBlkXp': 2,
'NGsBlkXp_units': 'Ry',
'GbndRnge': [1, 10],
'DysSolver': "n",
'QPkrange': [[1, 1, 8, 9]],
'DIP_CPU': "1 1 1",
'DIP_ROLEs': "k c v",
'X_CPU': "1 1 1 1",
'X_ROLEs': "q k c v",
'SE_CPU': "1 1 1",
'SE_ROLEs': "q qp b",
}
params_gw = Dict(dict=params_gw)
builder = YamboConvergence.get_builder()
##################scf+nscf part of the builder
builder.ywfl.scf.pw.structure = structure
builder.ywfl.scf.pw.parameters = parameter_scf
builder.kpoints = kpoints
builder.ywfl.scf.pw.metadata.options.max_wallclock_seconds = \
options['max_wallclock_seconds']
builder.ywfl.scf.pw.metadata.options.resources = \
dict = options['resources']
if 'queue_name' in options:
builder.ywfl.scf.pw.metadata.options.queue_name = options['queue_name']
if 'qos' in options:
builder.ywfl.scf.pw.metadata.options.qos = options['qos']
if 'account' in options:
builder.ywfl.scf.pw.metadata.options.account = options['account']
builder.ywfl.scf.pw.metadata.options.custom_scheduler_commands = options['custom_scheduler_commands']
builder.ywfl.nscf.pw.structure = builder.ywfl.scf.pw.structure
builder.ywfl.nscf.pw.parameters = parameter_nscf
builder.ywfl.nscf.pw.metadata = builder.ywfl.scf.pw.metadata
builder.ywfl.scf.pw.code = load_node(options['pwcode_pk'])
builder.ywfl.nscf.pw.code = load_node(options['pwcode_pk'])
builder.ywfl.scf.pw.pseudos = validate_and_prepare_pseudos_inputs(
builder.ywfl.scf.pw.structure, pseudo_family = Str(options['pseudo_family']))
builder.ywfl.nscf.pw.pseudos = builder.ywfl.scf.pw.pseudos
##################yambo part of the builder
builder.ywfl.yres.yambo.metadata.options.max_wallclock_seconds = \
options['max_wallclock_seconds']
builder.ywfl.yres.yambo.metadata.options.resources = \
dict = options['resources']
if 'queue_name' in options:
builder.ywfl.yres.yambo.metadata.options.queue_name = options['queue_name']
if 'qos' in options:
builder.ywfl.yres.yambo.metadata.options.qos = options['qos']
if 'account' in options:
builder.ywfl.yres.yambo.metadata.options.account = options['account']
builder.ywfl.yres.yambo.parameters = params_gw
builder.ywfl.yres.yambo.precode_parameters = Dict(dict={})
builder.ywfl.yres.yambo.settings = Dict(dict={'INITIALISE': False, 'COPY_DBS': False})
builder.ywfl.yres.max_iterations = Int(5)
builder.ywfl.yres.yambo.preprocessing_code = load_node(options['yamboprecode_pk'])
builder.ywfl.yres.yambo.code = load_node(options['yambocode_pk'])
builder.parent_folder = load_node(options['parent_pk']).outputs.remote_folder
builder.p2y = builder.ywfl
builder.precalc = builder.ywfl #for simplicity, to specify if PRE_CALC is True
builder.workflow_settings = Dict(dict={'type':'1D_convergence',
'what':'gap',
'where':[(1,8,1,9)],
'k-point':['Gamma'],
'PRE_CALC': False,})
#'what': 'single-levels','where':[(1,8),(1,9)]
var_to_conv = [{'var':['BndsRnXp','GbndRnge'],'delta': [[0,10],[0,10]], 'steps': 2, 'max_iterations': 3, \
'conv_thr': 0.2, 'conv_window': 2},
{'var':'NGsBlkXp','delta': 1, 'steps': 2, 'max_iterations': 3, \
'conv_thr': 0.2, 'conv_window': 2},
{'var':['BndsRnXp','GbndRnge'],'delta': [[0,10],[0,10]], 'steps': 2, 'max_iterations': 5, \
'conv_thr': 0.1, 'conv_window': 2},
{'var':'NGsBlkXp','delta': 1, 'steps': 2, 'max_iterations': 3, \
'conv_thr': 0.1, 'conv_window': 2},]
'''
{'var':'kpoints','delta': 1, 'steps': 2, 'max_iterations': 2, \
'conv_thr': 0.1, 'conv_window': 2, 'what':'gap','where':[(1,1)],}]
'''
for i in range(len(var_to_conv)):
print('{}-th variable will be {}'.format(i+1,var_to_conv[i]['var']))
builder.parameters_space = List(list = var_to_conv)
return builder
if __name__ == "__main__":
options = get_options()
builder = main(options)
running = submit(builder)
print("Submitted YamboConvergence; with pk=<{}>".format(running.pk))
As you can see, we have to provide workflow_settings, which encode some workflow logic:
{'type':'1D_convergence','what':'gap','where':[(k_v,vbM,k_c,cbm)],'where_in_words':['Gamma']})
The workflow submitted here looks for convergence on different parameters, searching each step a given parameter(1D). The iter is specified
with the input list parameters_space. This is a list of dictionaries, each one representing a given phase of the investigation. The quantity that tries
to converge is the gap(‘what’) between given bands evaluated at fixed k-points. It is possible to choose also and indirect gap(notice that,
changing the k-point mesh, the k-points will change index: for now we have not a logic to trace the index-change). The other functionality of the converge workflow is to converge single levels
(‘gap’->’single-levels’, [(k_v,vbM,k_c,cbm)]->[(k,b)]), useful in the study of molecules. It is possible also to search convergence simultaneously for
multiple gaps/levels, just adding tuples in the ‘where’ list. The workflow will take care of it and doesn’t stop until all the quantities are
converged(or the maximum restarts are reached).
The complete workflow will return the results of the convergence iterations, as well as a final converged calculation, from which we can parse the converged parameters, and a complete story of all the calculations of the workflow with all the information provided.
The data can be plotted using a function in :
from aiida_yambo.utils.plot_utilities import plot_conv
plot_conv(<workflow_pk>,title='Gap at Gamma for bulk hBN')
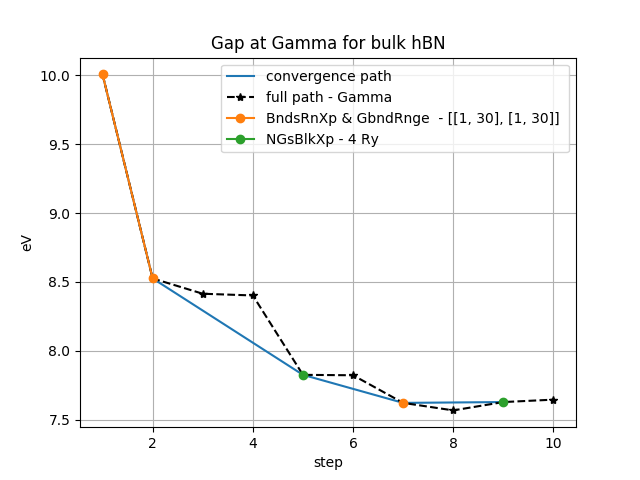
As you can see, the actual path of investigation is provided from inputs, but usually is always the same: bands and G-vectors cutoff (plus a final k-points convergence not seen here). The black stars represent overconverged results for each parameter-iteration. To see more examples, go to Plot results of YamboConvergence - ‘1D_convergence’. There, we can see real convergence on 2D-hBN. Please note that you have to set also builder.p2y and builder.pre_calc inputs, needed if p2y (parent is not yambo) and/or PRE_CALC (see workflow_settings) are needed.
Outputs of a YamboConvergence calculation are two: a Dict with the collection of information on all the calculations, and a Dict with the description of the last converged calculations, from which you can easily collect the converged parameters. To give a human-readable meaning to the output Dict, you can, after converted in dict (Dict is and AiiDA Data), build a pandas dataframe:
import pandas as pd
dataframe = pd.DataFrame(outdict)